What Would Happen to Someone Treated With a Drug That Inhibited the Release of Bile Salts?
Introduction
Inhibition of the bile common salt export pump (BSEP) past a drug has been implicated as a gamble factor for the drug's potential to crusade drug-induced liver injury (DILI) (Dawson et al., 2011; Morgan et al., 2013). Several high-profile DILI-causing drugs, for example troglitazone (Funk et al., 2001; Smith, 2003), bosentan (Fattinger et al., 2001; Eriksson et al., 2011), and nefazodone (Kostrubsky et al., 2006), have been shown to be BSEP inhibitors. Furthermore, prediction of toxicity by these molecules has been uneven; for case, neither bosentan nor troglitazone displayed toxicity in fauna models (Leslie et al., 2007; Lauer et al., 2009). Predictions involving hepatobiliary transporter IC50 values in in vitro assays have shown amend predictive ability (Dawson et al., 2011; Morgan et al., 2013). Improving the ability to predict the frequency and severity of DILI with BSEP inhibitors volition let those involved in drug development to better judge the run a risk involved with moving a drug in development into the clinic or beyond early on-stage clinical trials.
DILIsym® is a multi-calibration mechanistic model incorporating numerous functions of the liver and disruptions of the function with the goal of predicting the DILI potential of drugs at various stages in the development procedure (Howell et al., 2012, 2014; Woodhead et al., 2012; Shoda et al., 2014). Previously, we accept synthetic and validated a model of bile acid homeostasis and transporter inhibition within DILIsym® (Woodhead et al., 2014). Nosotros establish that inhibiting BSEP could lead to significant increases in bile acid concentrations in the liver, and that the effects of bile acrid transporter inhibitors should be considered on a simulated population equally well as on a single baseline individual. We have expanded that model to include a representation of bile acid-mediated toxicity based on experiments performed by Yang et al. (2013). In that work, we synthetic a relationship betwixt intracellular bile acid concentration and cellular ATP. We used this relationship to predict cellular necrosis using the existing human relationship between ATP and cell death in DILIsym®. This model has been used previously to finer predict the frequency and timing of ALT elevations observed with troglitazone in clinical trials, and has predicted the departure between troglitazone and the like non-toxic drug pioglitazone (Yang et al., 2014).
In the nowadays work, we use this model of bile acid-mediated cytotoxicity to model the observed hepatotoxicity of bosentan, telmisartan, and CP-724,714. Bosentan is a currently marketed medication for pulmonary arterial hypertension that carries a black-box warning for hepatotoxicity. In clinical trials a dose of thousand mg/day of bosentan caused between 8 and 18% of individuals to feel increases in serum ALT greater than 3-fold (Fattinger et al., 2001). Bosentan has too been shown to be a relatively potent BSEP inhibitor. Telmisartan, an angiotensin Two receptor agonist used in hypertension treatment, is also a relatively stiff BSEP inhibitor, just has non been reported to cause hepatotoxicity in humans (Morgan et al., 2013). CP-724,714 is an anti-cancer drug that was terminated from development later Stage 2 clinical trials revealed liver signals (Guo et al., 2008). CP-724,714 has been shown to inhibit multiple transporters, including BSEP, in addition to being a mitochondrial toxin (Feng et al., 2009). In this report we explore the DILIsym® software's (version 2C) predictions for the toxicity of bosentan in humans, and the lack of toxicity of bosentan in rats and telmisartan in humans. We volition also test several theories well-nigh the toxicity of CP-724,714 in order to propose potential viable explanations for the observed toxicity in early on clinical trials.
Methods
The construction and validation of the bile acid homeostasis model in DILIsym® is described in a previous newspaper (Woodhead et al., 2014). DILIsym® models the synthesis and enterohepatic recirculation of two master potentially toxic bile acids, chenodeoxycholic acid (CDCA) and lithocholic acid (LCA), and their amide (and sulfate in the instance of LCA) conjugates. DILIsym® describes the intrahepatic accumulation of these toxic bile acids as well equally the concentrations in the gallbladder, portal blood, gut lumen, and systemic blood. Concentrations of bile acid transporter inhibitors are modeled using a physiologically-based pharmacokinetic model (PBPK) described in depth in previous papers (Howell et al., 2012; Woodhead et al., 2012). The bile acrid concentrations are linked to ATP decline every bit described below; the effect of ATP decline on eventual hepatocyte necrosis is described by a model of the hepatocyte life wheel too described in previous papers (Howell et al., 2012; Woodhead et al., 2012; Shoda et al., 2014). A diagram of the interaction between the diverse submodels of DILIsym® employed in this paper can be seen in Figure 1.
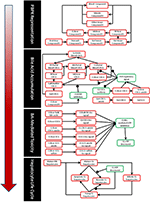
Figure 1. Partial diagram of the portions of DILIsym® used for this newspaper, including the PBPK representation, representation of bile acrid homeostasis and aggregating, link between bile acid concentrations and toxicity, and the hepatocyte life wheel.
The DILIsym® model of bile acid toxicity is based on in vitro experiments comparing the intracellular level of LCA and CDCA to cellular ATP levels (Yang et al., 2013). In society to construct the connectedness betwixt bile acid accumulation and toxicity, a pocket-size model of the DILIsym® ATP turnover model was synthetic (Yang et al., 2013); a diagram of this model is shown in Effigy two. The relationship between intracellular ATP and intracellular bile acids was modeled by the post-obit equation:
where k usage is the rate of ATP usage in the cell, k synth is the rate of ATP synthesis in the cell, and Due south is the signal for ATP synthesis inhibition by bile acids. South has Hill blazon behavior and is given by the post-obit equation:
where V max,S is the maximum denominator for the signal, K 1000,Southward is the Michaelis constant for the signal, and H is the Loma coefficient for the betoken. [BA] filibuster is the delayed intracellular bile acid concentration, given by the standard delay equation:
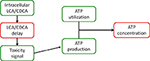
Figure 2. Diagram of the model of ATP turnover used to parameterize the hepatocellular toxic response to accumulation of bile acids. The model was prepare to accept the experimentally measured intracellular concentration of each bile acid, and parameters were fit to the model in order to fit the experimentally observed intracellular ATP levels for each bile acid exposure.
The filibuster constant τ, V max,S , Grand m,S , and H were the parameters that were fitted to the ATP time course from the experiment. These parameters were and then applied to the intracellular bile acrid concentrations modeled in DILIsym®. We applied the parameters for the unconjugated bile acids measured in the experiments to the conjugated bile acids in DILIsym®; for instance, nosotros used the unconjugated LCA parameters to depict the toxicity of LCA amide and sulfate conjugates. While some data exist describing the relationship between intracellular amide- and sulfate-conjugated bile acids and toxicity (Chatterjee et al., 2013), these are not enough to justify using unlike toxicity parameters for conjugated and unconjugated bile acids; this is an area of potential refinement for DILIsym®.
Physiologically-based pharmacokinetic (PBPK) models were constructed for bosentan, telmisartan, and CP-724,714 in humans using available in vivo time course data (Weber et al., 1996; Stangier et al., 2000; Munster et al., 2007). For simulations where bosentan metabolism was impaired, the V max for the metabolism of both metabolites was represented as x% of their baseline value. Both bosentan and telmisartan are active transport substrates; the active transport parameters for both were adapted for use in DILIsym® from Ménochet et al. (2012). The bosentan PBPK model includes the major and pocket-sized metabolite, with metabolism parameters based on published in vitro metabolism information (Ubeaud et al., 1995); these were included because the pocket-size metabolite of bosentan also inhibits BSEP (Fattinger et al., 2001). The major metabolite of telmisartan, a glucuronide, has not been shown to inhibit BSEP and and so was non included in the PBPK model. CP-724,714's circuitous metabolism was not modeled explicitly; nonetheless, a single metabolite compartment in the liver was included in society to test the hypothesis of whether metabolite accumulation could explicate CP-724,714 toxicity. PBPK model results for human bosentan are shown in Effigy three; those for man telmisartan are shown in Figure 4; and those for homo CP-724,714 in Figure 5. For bosentan in rats, data did not be in the literature. Information provided to us from Amgen, Inc. (Thou Oaks, CA) were used to parameterize the bosentan rat PBPK model shown in Figure half-dozen.
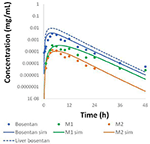
Figure 3. Fake plasma concentration of bosentan in humans and its two metabolites after a 500 mg oral dose compared to information from Weber et al. (1996).
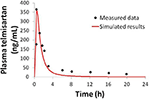
Figure four. Fake plasma telmisartan concentration in humans after a single 80 mg oral dose compared to data from Stangier et al. (2000).

Figure 5. Imitation plasma human concentrations of CP-724,714 after an oral dose of 250 mg (A) and 400 mg (B) compared to data from Munster et al. (2007). The 250 mg dose data represented in this figure are a compilation of the results from the get-go dose of several dissimilar dosing protocols.
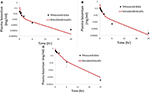
Figure half dozen. Simulated plasma bosentan concentrations in rats later an intravenous oral dose of ten mg (A), 30 mg (B), and 100 mg (C) compared to information provided by Amgen, Inc. (One thousand Oaks, CA).
Inhibition constants for bosentan were taken from published research that found K i values for both bosentan (12 μM) and the minor metabolite of bosentan (8.five μM) (Fattinger et al., 2001). The inhibition of BSEP by bosentan and its metabolite were modeled every bit noncompetitive in agreement with the data. These information are from rat Bsep vesicles; we assigned the aforementioned Chiliad i value to humans, as there is testify suggesting that rat Bsep and human BSEP are inhibited with similar potency by bosentan (Mano et al., 2007). Bile acrid uptake inhibition constants for bosentan were taken from work done past Leslie et al. (2007). The telmisartan K i was approximated using the IC50 value reported in the literature (Morgan et al., 2013). A list of K i values used in our simulations is included in Tabular array i. In addition, bosentan is a strong inducer of its own uptake into the liver and metabolism (Dingemanse and van Giersbergen, 2004); these effects are included in our PBPK model. It is important to note that for each drug, only the parameters related to drug pharmacokinetics and transporter inhibition were changed; all other remaining parameters were not changed from drug to drug.

Tabular array one. Inhibition constants used for the compounds in the simulations conducted for this paper.
For CP-724,714 ICfifty values, experiments were run in rat Bsep, dog Bsep, and homo BSEP-expressing vesicles. CP-724,714 was synthesized past Pfizer, Inc. (Groton, CT), while radiochemicals were purchased from Perkin Elmer (Perkin Elmer, Waltham MA). Other chemicals were purchased from Sigma-Aldrich (Sigma-Aldrich, St Louis, MO) or were of belittling grade. Membrane vesicles were obtained from Sf9 cells non expressing and expressing human BSEP (Solvo Biotechnology, Szeged, Hungary), rat Bsep (AB Life Technologies, Waltham, MA), or domestic dog Bsep (AB Life Technologies).
CP-724,714 was incubated (2 or 5 min) with membrane vesicle preparations (total poly peptide: fifty μg/well) and the probe substrate, taurocholate (ii μM). Serial dilutions of CP-724,714 (30 mM stock, 10 mM, three.16 mM, 1 mM, 316 μM, 100 μM, 31.half-dozen μM, 10 μM) were prepared in DMSO. Incubations in duplicate were carried out in the absenteeism or presence of 4 mM ATP to distinguish between transporter mediated uptake and passive improvidence into the vesicles. CP-724,714 was added to the reaction mixture in 0.75 μl of solvent (1% of the final incubation volume). Glyburide (100 – 0.1 μM) was used as the positive control inhibitor. Reaction mixtures were pre-incubated for 10 min at 37°C. Reactions were started by the addition of 25 μl of 12 mM MgATP (or AMP, as disodium salt, for groundwork controls), pre-incubated separately. Reactions were stopped by the addition of 200 μl of ice-cold washing buffer and immediate filtration via glass fiber filters mounted to a 96-well plate (filter plate). The filters were done, stale and the amount of substrate inside the filtered vesicles adamant past liquid scintillation. Maximal observed inhibition ranged between 94.8 and 99.viii% for CP-724,714. Farther data on this method can be found in the literature (Kis et al., 2009).
Bosentan dosing in humans was fake every bit a 500-mg twice-daily dose for 30 days. The doses were given in one case every 12 h; this is i of the dosing regimens in the clinical trials that demonstrated toxicity as reported by Fattinger et al. (2001). Telmisartan dosing was false as a 50, 3000, or 12,000-mg dose one time daily for 30 days; common clinical dosing regimens range from twoscore to 80 mg per twenty-four hour period (Meredith, 1999). CP-724,714 was dosed three times daily, or once every eight h. A range of CP-724,714 doses were simulated in order to comprehend the range of exposure levels reported by Guo et al. (2008). Three simulated meals per day were included in all human simulations, and simulated to occur concurrent with drug dosing. Bosentan dosing in rats was simulated as a fifty mg/kg dose once daily.
DILIsym® contains simulated populations, or SimPops™, that represent a plausible range of variability in primal model parameters. The human SimPops™ and rat SimPops™ used in the course of these simulations comprise variability in several bile acrid send parameters and are described in our previous work (Woodhead et al., 2014; Yang et al., 2014). At that place are 331 individuals in the man SimPops™ and 191 individuals in the rat SimPops™; although the numbers of individuals are different, both the homo and the rat SimPops™ are designed to business relationship for the entire plausible range in variability in bile acrid transport parameters and bile acid profiles observed in a sample population (Woodhead et al., 2014). Pharmacokinetic variability in bosentan disposition was too included in one SimPops™ simulation; for this population, a normal distribution around four variables (oral absorption coefficient, liver uptake V max, and major and minor metabolite formation V max) was superimposed upon the existing small-scale human being SimPops™. The range of these parameter values was based on a known range of variability for hepatobiliary transporters (Meier et al., 2006) and upon previous simulations where pharmacokinetic variability was considered (Woodhead et al., 2012).
Results
Results from the SimPops™ analysis with bosentan are presented in Table 2. When we false bosentan in the human SimPops™ using the baseline assumptions (noncompetitive inhibition, no basolateral inhibition, normal metabolism), 1 individual out of 331 adult ALT elevations greater than 3-fold above the baseline value. When potential variability in pharmacokinetics was incorporated into the SimPops™, the predicted incidence rate rose to 3 out of 331. While this successfully predicts the potential toxicity of bosentan, the incidence charge per unit is well below the incidence charge per unit of eight–18% observed during the clinical trials (Fattinger et al., 2001).

Tabular array 2. SimPops™ simulation results for simulated ALT elevations acquired past bosentan and telmisartan.
A genetic polymorphism that limits CYP metabolism has been shown to correlate with an increased rate of toxicity from bosentan exposure (Markova et al., 2013). We have simulated this example using the human SimPops™ by decreasing the bosentan metabolism V max values for both minor and major metabolites by 10-fold. This change led to eight simulated individuals out of 331 (2.42%) developing ALT elevations. DILIsym® correctly predicts the increased risk of toxicity due to this potential genetic polymorphism, though the predicted incidence rate is still less than the overall rate in the general population from the clinical study.
When bosentan was likewise simulated in the rat SimPops™, zilch individual rats out of 191 developed ALT elevations. This is consistent with preclinical observations which have reported no toxicity in the rat (Leslie et al., 2007). It has been suggested that the divergence in bile acid uptake inhibition by bosentan between rats and humans contributes to the species divergence in hepatotoxicity (Ansede et al., 2010). In order to test this theory, we ran a simulation in the rat SimPops™ with uptake inhibition eliminated (K i set to 1 × 10x); though bile acrid accumulation in the liver was predicted (results not shown), no toxicity was observed. This suggests that the difference in the bile acrid pool between humans and rats is likely a more than pregnant correspondent to the species deviation in toxicity.
In gild to ensure that the model can differentiate non-toxic BSEP inhibitors from toxic BSEP inhibitors, we modeled telmisartan in the human being SimPops™. No toxicity was predicted in the human being SimPops™, which is consequent with clinical observations. This was true whether telmisartan was modeled as a competitive or a noncompetitive inhibitor.
While in that location is scant difference between predicting one individual developing toxicity and predicting nothing individuals developing toxicity (out of 331), a fuller agreement of the risk of a given chemical compound can be gained by investigating more mechanistic information within DILIsym®. Figure 7 displays the minimum hepatic ATP for each private in the human and rat SimPops™ for bosentan and the homo SimPops™ for telmisartan (modeled as both a competitive and not-competitive inhibitor of BSEP). We can see that while merely one simulated private in the human SimPops™ had an ATP decline subsequently bosentan dosing that ultimately led to toxicity, many more had ATP reductions visibly beneath the baseline value. This was non truthful of the rat, or of telmisartan in either example; the faux individuals all accept hepatic ATP values very close to the baseline value. This places the departure betwixt bosentan and telmisartan, and the deviation between rat and homo, in sharper relief and demonstrates the potential of the model for predicting species differences in toxicity and differentiating betwixt toxic and not-toxic BSEP inhibitors.
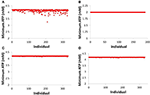
Effigy 7. ATP levels in individuals after faux dosing of bosentan to the human SimPops™ (A) and the rat SimPops™ (B); and with simulated telmisartan dosing to the human SimPops™ when represented as a competitive (C) and noncompetitive (D) inhibitor. Each point represents an individual within the man SimPops™; the baseline ATP concentration in humans is iv.2 mM, while in rats the baseline ATP concentration is 2.0 mM.
The simulation results for CP-724,714 in the baseline human individual, and a comparing of these results to the clinical observations in Guo et al. (2008), are shown in Figure 8. The graph compares the normalized liver function exam (LFT) elevation against the AUC of the drug in the patient's bloodstream on Day 22 (Day 1 of Cycle ii) of drug dosing, a measure of steady-state drug exposure. The normalized LFT elevation used by the Guo et al. (2008) paper to which our simulations are compared is given past the following expression (Guo et al., 2008):

Figure 8. Normalized LFT elevation at increasing exposure equally measured by AUC0-24 on Day i of Cycle two (Day 22 overall) in the baseline human in DILIsym® and the clinical trial patients from Guo et al. (2008). The lines refer to the dose response simulated in DILIsym® under various assumed conditions listed in the table legend. "Baseline assumptions" refers to the instance where merely the parent CP-724,714 inhibits BSEP competitively and does not inhibit the electron transport chain (ETC). "Metabolite inhibition" refers to the instance where the metabolite of CP-724,714 inhibits BSEP with the aforementioned K i as the parent compound. "Poor clearance" refers to the case where the metabolite biliary clearance value is set to a value 10-fold lower than the parent biliary clearance value. "Noncompetitive" refers to the example where all BSEP inhibition is modeled as noncompetitive. "Added ETC inhibition" refers to the case where inhibition of the electron transport chain was simulated for both parent and metabolite. The points are individual patients from Guo et al. (2008). The purple dashes are the individuals in a human SimPops™.
The simulation results are the maximum normalized LFT height at each dose faux for each individual case. The graph shows that bile acid inhibition alone cannot explicate the clinical toxicity, equally liver function tests did non elevate in the simulations when bile acid inhibition alone was faux. This was true for both competitive and noncompetitive inhibition. All the same, the inclusion of mitochondrial toxicity did, in fact, pb to a toxic response that was augmented when bile acid aggregating also occurred. The data are best described here by this combination of competitive inhibition and electron transport chain (ETC) inhibition.
The simulations besides predict that both BSEP and mitochondrial ETC inhibition by CP-724,714'due south major metabolite are necessary to explain the toxicity, since CP-724,714 is rather rapidly metabolized by the liver and then parent residence fourth dimension within the liver is somewhat limited. When the potential activeness of the metabolite is non included, no toxicity is shown no matter the machinery or combination of mechanisms selected. The accumulation of this metabolite is as well necessary to explain the toxicity; a biliary clearance of 10% of the value for the parent chemical compound was used in order to reproduce the clinical toxicity.
SimPops™ results for CP-724,714 in humans are also shown in Figure 8, with each private in the SimPops™ represented by a purple dash. We found that while the fake baseline individual did not brandish the toxicity that would have been expected from the clinical data, the population sample did contain several individuals who developed clinically-relevant ALT elevations, but only if the BSEP inhibition was noncompetitive. Furthermore, the range of injury in the SimPops™ is far wider than the range reported in the clinical study; the well-nigh astringent normalized LFT elevation in our simulated population was half dozen.5, while the largest clinical normalized LFT in that exposure range was about two. Twelve individuals in our 331-individual SimPops™, or 3.62%, developed toxicity; this is lower than the 36% of individuals in the exposure range near the simulated dose who developed LFT elevations. No individuals in the SimPops™ adult toxicity if the inhibition was competitive, demonstrating the importance of differentiating betwixt modes of inhibition when determining BSEP inhibition constants.
Uncertainty about the biliary clearance of the CP-724,714 metabolite has an impact on faux ALT elevations. Figure 9 shows imitation ALT elevations at a constant dose of CP-724,714 when the biliary clearance of the metabolite is modulated. This variable does not touch on the plasma pharmacokinetics of parent CP-724,714; it is thus a degree of freedom in DILIsym®. However, as Figure 9 shows, it has a significant effect on the predicted toxicity of CP-724,714; DILIsym® suggests, therefore, that experiments clarifying the amount of hepatic accumulation and clearance of CP-724,714's major metabolite should exist conducted in order to fully elucidate the molecule's toxicity.
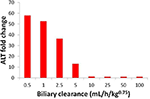
Figure nine. ALT elevations at unlike values of metabolite biliary clearance given competitive BSEP inhibition and ETC inhibition. The "poor clearance" cases explored in Effigy 8 are represented here past the biliary clearance value of 5 mL/h/kg0.75.
Discussion
Nosotros have used DILIsym® to model bosentan and found that, using the mechanistic simulation results calculating ATP decline, DILIsym® suggests that the potential for toxicity in a human population is greater than that in a rat population. Furthermore, the same ATP turn down simulation results bear witness a clear difference between the human being population response to bosentan and to telmisartan. While the prediction of the toxic potential of bosentan was well below the bodily clinical incidence charge per unit, these results even so testify hope for the ability to use a mechanistic mathematical model of DILI, rather than pocket-size-animal models, to predict the human being toxicity of a BSEP inhibitor. More than revealingly, the modeling do suggested a potential reason for this discrepancy in toxicity between rats and humans. Previous work in this area demonstrated the deviation in uptake transporter inhibition betwixt rats and humans (Leslie et al., 2007; Ansede et al., 2010) and proposed that this was a contributing factor to the species departure. Nevertheless, our simulations suggest that removing the uptake inhibition from the rat model altogether does not lead to liver toxicity in the rat. This suggests that the difference in bile acid pools and metabolic pathways are well-nigh likely the strongest contributor to the species difference in toxicity. The rat bile acid puddle has more non-toxic bile acids, such as cholic acrid and muricholic acrid, and less of the toxic bile acids CDCA and LCA than the human (Hofmann, 2009; García-Cañaveras et al., 2012). Furthermore, the rat can hydroxylate LCA into the less-toxic hyodeoxycholic acid, which is a pathway that does non occur in the homo (Hofmann, 2004). Considering of these species differences in bile acid metabolism, our modeling questions the utility of current pocket-size-animal models in the prediction of BSEP inhibitor-mediated toxicity in humans.
Our results comparing the ATP decrement in the liver caused by bosentan and telmisartan demonstrate that DILIsym® can differentiate between a toxic and a non-toxic chemical compound with a similar IC50. In the case of bosentan and telmisartan, the difference is by and large a pharmacokinetic one; telmisartan is dosed at a far lower dose than is bosentan. However, our results with increasing telmisartan dose suggest that a much higher dose of telmisartan, compared to bosentan, is necessary to crusade toxicity in a human SimPops™. The suggestion is that dose is not necessarily predictive on its own; differences in metabolism and in liver uptake transporter affinity and chapters both are likely contributors to the differences in toxicity between bosentan and telmisartan.
While nosotros qualitatively predict the species difference in bosentan toxicity and the toxicity of bosentan compared with the safety of telmisartan, we substantially underpredict the incidence rate of the toxicity of bosentan in the general population. Clinical trials predict 8–18% toxicity at the 1000 mg/solar day dosing level; DILIsym® predicted a much lower incidence rate even when individuals with metabolic polymorphisms were considered.
There are several possible reasons for this discrepancy, by and large related to the possibility of other mechanisms of toxicity not currently included in the DILIsym® model of bosentan. First, we do not represent the inhibition of basolateral bile acrid ship by bosentan; this could forestall a potential clearance pathway for toxic bile acid species and pb to more toxicity. Contempo research has shown that bosentan has the potential to inhibit MRP4, a basolateral bile acid transporter (Morgan et al., 2013). Exploratory simulations representing bosentan as a basolateral transporter inhibitor (reported in Table 2) suggest that this caption is likely only part of the problem; if the bosentan basolateral transporter Yard i is particularly depression, nevertheless, this could business relationship for some of the discrepancy in predicted incidence rates.
Second, DILIsym® does not represent bile acid toxicity in a mechanistic manner. Recent research suggests that the bile acids affect the mitochondria and potentially lead to the mitochondrial membrane permeability transition (Schulz et al., 2013); representing this effect in DILIsym® could introduce extra sources of variability and could lead to a higher predicted toxicity incidence rate. Indeed, work is underway in this surface area for DILIsym® and preliminary results suggest that the mechanistic model increases the predicted rate of bosentan toxicity without predicting toxicity in the rat; this work volition be the focus of a subsequent paper.
Third, bosentan could cause toxicity through mechanisms not currently included in the model for bosentan or not currently represented in DILIsym®. Bosentan has been shown to inhibit phospholipid transport in rodents (Fouassier et al., 2002). Phospholipids protect the bile duct from the cytotoxic effect of bile acids; inhibition of phospholipid transport has been shown to lead to liver toxicity in the case of itraconazole (Yoshikado et al., 2011). While bosentan'due south mechanism of action is unlike to that of itraconazole, this shows that the power of bosentan to interfere with phospholipid transport could be at to the lowest degree partially responsible for the observed toxicity of bosentan. Phospholipid transport and the toxic effect of its inhibition are non currently represented in DILIsym®; this is an area for potential future research and refinement.
Also, bosentan's effects on the mitochondria take not been elucidated. Previous enquiry has shown that at that place is a correlation between the power to inhibit BSEP and toxic effects on the mitochondria, and that this combined effect is itself correlated with DILI gamble (Aleo et al., 2014). Furthermore, our simulations with CP-724,714 demonstrated that the toxicity seen in the clinic could non exist explained without representing the drug's mitochondrial furnishings; while our research suggests that information technology is plausible that a chemical compound could cause toxicity through bile acid transporter inhibition lone, it is as well plausible that the toxicity of bosentan is due to a combined mitochondria/bile acrid toxicity mechanism. Indeed, our modeling showed that this was near probable the case with CP-724,714. Enquiry in this area is underway in our grouping and will focus on potential synergy between mitochondrial toxicity and bile acid buildup in the liver.
Fourth, DILIsym® does non stand for the intracellular trafficking and potential localization of the drug within the hepatocyte. Further research in this area could help usa ameliorate DILIsym® and thus our prediction of toxicity incidence rates.
Our simulations with CP-724,714 demonstrate the ability of mechanistic modeling to consider and prioritize multiple hypotheses when modeling compounds where the cause of toxicity is non fully established. Potential variability in the caste of hepatic accumulation of a compound that inhibits BSEP is particularly important, as shown past Figure 9. This is an especially salient point to consider given that the toxicity of compounds such as troglitazone are suspected to be due to the hepatic accumulation of a BSEP-inhibiting metabolite (Masubuchi, 2006). Furthermore, the deviation between competitive and non-competitive inhibition, the potential contribution of ETC inhibition to the observed toxicity, and the toxic potential of CP-724,714'southward metabolites are all sources of doubtfulness that required a hypothesis-based approach to modeling. While this is of limited value for predicting toxicity on its own, the hypothesis-based modeling arroyo is potentially valuable in determining which experiments would be nigh impactful in using the model to properly predict toxicity. In the case of CP-724,714, these experiments include hepatic accumulation studies, BSEP inhibition studies, and ETC inhibition studies on CP-724,714's metabolites.
Conflict of Interest Statement
The authors declare that the inquiry was conducted in the absence of whatsoever commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to acknowledge Yurong Lai (Pfizer) for help with the BSEP inhibition studies, Michael D. Aleo and Denise Robinson-Gravatt for discussions around DILIsym® implementation for CP-724,714, and the product data management and projection squad scientists whose CP-724,714 data facilitated this modeling effort. We would also similar to acknowledge Ryan Morgan (Amgen, Inc.) for many helpful discussions and assistance with the management of this inquiry. We also acknowledge Ryan Morgan, Chuck Qualls, Hisham Hamadeh, Sabina Buntich, Nataraj Kalyanaraman and Dean Hickman (Amgen) and Andy Mould and Jim Howard (Covance Laboratories) for their work on the bosentan pharmacokinetic rat studies that take been used to optimize the rat bosentan model.
References
Ansede, J. H., Smith, W. R., Perry, C. H., St Claire, R. 50. tertiary., and Brouwer, G. R. (2010). An in vitro analysis to assess transporter-based cholestatic hepatotoxicity using sandwich-cultured rat hepatocytes. Drug Metab. Dispos. Biol. Fate Chem. 38, 276–280. doi: ten.1124/dmd.109.028407
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Chatterjee, Southward., Bijsmans, I. T. One thousand. W., van Mil, S. W. C., Augustijns, P., and Annaert, P. (2013). Toxicity and intracellular accumulation of bile acids in sandwich-cultured rat hepatocytes: role of glycine conjugates. Toxicol. Vitro Int. J. Publ. Assoc. 28, 218–230. doi: 10.1016/j.tiv.2013.10.020
Pubmed Abstract | Pubmed Total Text | CrossRef Total Text | Google Scholar
Dawson, S. E., Stahl, S., Paul, Northward., Barber, J., and Kenna, J. M. Thousand. (2011). In vitro inhibition of the bile salt export pump correlates with chance of cholestatic drug induced liver injury in man. Drug Metab. Dispos. Biol. Fate Chem. forty, 130–138. doi: 10.1124/dmd.111.040758
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text
Eriksson, C., Gustavsson, A., Kronvall, T., and Tysk, C. (2011). Hepatotoxicity by bosentan in a patient with portopulmonary hypertension: a example-written report and review of the literature. J. Gastrointest. Liver Dis. 20, 77–80.
Pubmed Abstruse | Pubmed Total Text | Google Scholar
Fattinger, 1000., Funk, C., Pantze, M., Weber, C., Reichen, J., Stieger, B., et al. (2001). The endothelin adversary bosentan inhibits the canalicular bile common salt export pump: a potential mechanism for hepatic adverse reactions. Clin. Pharmacol. Ther. 69, 223–231. doi: x.1067/mcp.2001.114667
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Feng, B., Xu, J. J., Bi, Y.-A., Mireles, R., Davidson, R., Duignan, D. B., et al. (2009). Function of hepatic transporters in the disposition and hepatotoxicity of a HER2 tyrosine kinase inhibitor CP-724,714. Toxicol. Sci. 108, 492–500. doi: x.1093/toxsci/kfp033
Pubmed Abstract | Pubmed Total Text | CrossRef Full Text | Google Scholar
Fouassier, L., Kinnman, N., Lefèvre, G., Lasnier, East., Rey, C., Poupon, R., et al. (2002). Contribution of mrp2 in alterations of canalicular bile germination by the endothelin antagonist bosentan. J. Hepatol. 37, 184–191. doi: ten.1016/S0168-8278(02)00107-1
Pubmed Abstract | Pubmed Total Text | CrossRef Full Text | Google Scholar
Funk, C., Pantze, Chiliad., Jehle, L., Ponelle, C., Scheuermann, G., Lazendic, M., et al. (2001). Troglitazone-induced intrahepatic cholestasis by an interference with the hepatobiliary export of bile acids in male person and female person rats. Correlation with the gender divergence in troglitazone sulfate formation and the inhibition of the canalicular bile salt export pump (Bsep) by troglitazone and troglitazone sulfate. Toxicology 167, 83–98. doi: 10.1016/S0300-483X(01)00460-7
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
García-Cañaveras, J. C., Donato, 1000. T., Castell, J. 5., and Lahoz, A. (2012). Targeted profiling of circulating and hepatic bile acids in human, mouse, and rat using a UPLC-MRM-MS-validated method. J. Lipid Res. 53, 2231–2241. doi: 10.1194/jlr.D028803
Pubmed Abstract | Pubmed Full Text | CrossRef Total Text | Google Scholar
Guo, F., Letrent, S. P., Munster, P. Due north., Britten, C. D., Gelmon, K., Tolcher, A. W., et al. (2008). Pharmacokinetics of a HER2 tyrosine kinase inhibitor CP-724,714 in patients with advanced cancerous HER2 positive solid tumors: correlations with clinical characteristics and condom. Cancer Chemother. Pharmacol. 62, 97–109. doi: 10.1007/s00280-007-0579-four
Pubmed Abstruse | Pubmed Total Text | CrossRef Full Text | Google Scholar
Howell, B. A., Siler, S. Q., Shoda, L. K. Yard., Yang, Y., Woodhead, J. L., and Watkins, P. B. (2014). A mechanistic model of drug-induced liver injury AIDS the interpretation of elevated liver transaminase levels in a stage I clinical trial. CPT Pharmacomet. Syst. Pharmacol. iii, e98. doi: 10.1038/psp.2013.74
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Howell, B. A., Yang, Y., Kumar, R., Woodhead, J. L., Harrill, A. H., Clewell, H. J. 3rd, et al. (2012). In vitro to in vivo extrapolation and species response comparisons for drug-induced liver injury (DILI) using DILIsym™: a mechanistic, mathematical model of DILI. J. Pharmacokinet. Pharmacodyn. 39, 527–541. doi: x.1007/s10928-012-9266-0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kis, Due east., Ioja, E., Nagy, T., Szente, L., Herédi-Szabó, K., and Krajcsi, P. (2009). Issue of membrane cholesterol on BSEP/Bsep action: species specificity studies for substrates and inhibitors. Drug Metab. Dispos. Biol. Fate Chem. 37, 1878–1886. doi: ten.1124/dmd.108.024778
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kostrubsky, S. E., Strom, Southward. C., Kalgutkar, A. S., Kulkarni, Due south., Atherton, J., Mireles, R., et al. (2006). Inhibition of hepatobiliary ship as a predictive method for clinical hepatotoxicity of nefazodone. Toxicol. Sci. 90, 451–459. doi: 10.1093/toxsci/kfj095
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Leslie, Due east. G., Watkins, P. B., Kim, R. B., and Brouwer, Grand. 50. R. (2007). Differential inhibition of rat and homo Na+-dependent taurocholate cotransporting polypeptide (NTCP/SLC10A1)by bosentan: a mechanism for species differences in hepatotoxicity. J. Pharmacol. Exp. Ther. 321, 1170–1178. doi: x.1124/jpet.106.119073
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Markova, S. M., De Marco, T., Bendjilali, N., Kobashigawa, East. A., Mefford, J., Sodhi, J., et al. (2013). Association of CYP2C9*two with bosentan-induced liver injury. Clin. Pharmacol. Ther. 94, 678–686. doi: 10.1038/clpt.2013.143
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Meier, Y., Pauli-Magnus, C., Zanger, U. K., Klein, Grand., Schaeffeler, E., Nussler, A. One thousand., et al. (2006). Interindividual variability of canalicular ATP-binding-cassette (ABC)-transporter expression in human liver. Hepatol. 44, 62–74. doi: ten.1002/hep.21214
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Morgan, R. E., van Staden, C. J., Chen, Y., Kalyanaraman, N., Kalanzi, J., Dunn, R. T., et al. (2013). A multifactorial approach to hepatobiliary transporter cess enables improved therapeutic chemical compound development. Toxicol. Sci. 136, 216–241. doi: 10.1093/toxsci/kft176
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Munster, P. N., Britten, C. D., Mita, K., Gelmon, G., Minton, South. Due east., Moulder, S., et al. (2007). First report of the safe, tolerability, and pharmacokinetics of CP-724,714 in patients with avant-garde malignant solid HER2-expressing tumors. Clin. Cancer Res. thirteen, 1238–1245. doi: 10.1158/1078-0432.CCR-06-1539
Pubmed Abstract | Pubmed Total Text | CrossRef Full Text | Google Scholar
Schulz, South., Schmitt, Southward., Wimmer, R., Aichler, 1000., Eisenhofer, S., Lichtmannegger, J., et al. (2013). Progressive stages of mitochondrial devastation caused by cell toxic bile salts. Biochim. Biophys. Acta 1828, 2121–2133. doi: ten.1016/j.bbamem.2013.05.007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Shoda, L. K. Thou., Woodhead, J. 50., Siler, South. Q., Watkins, P. B., and Howell, B. A. (2014). Linking physiology to toxicity using DILIsym(®), a mechanistic mathematical model of drug-induced liver injury. Biopharm. Drug Dispos. 35, 33–49. doi: 10.1002/bdd.1878
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Stangier, J., Su, C. A., and Roth, Westward. (2000). Pharmacokinetics of orally and intravenously administered telmisartan in healthy young and elderly volunteers and in hypertensive patients. J. Int. Med. Res. 28, 149–167. doi: 10.1177/147323000002800401
Pubmed Abstract | Pubmed Full Text | CrossRef Total Text | Google Scholar
Ubeaud, M., Schmitt, C., and Jaeck, D. (1995). Bosentan, a new endothelin receptor adversary: prediction of the systemic plasma clearance in man from combined in vivo and in vitro data. Xenobiotica 25, 1381–1390. doi: 10.3109/00498259509061925
Pubmed Abstruse | Pubmed Total Text | CrossRef Full Text | Google Scholar
Weber, C., Schmitt, R., Birnboeck, H., Hopfgartner, G., Marle, S. P. 5., and Jones, C. (1996). Pharmacokinetics and pharmacodynamics of the endothelin-receptor antagonist bosentan in healthy human subjects. Clin. Pharmacol. Ther. 60, 124–137. doi: 10.1016/S0009-9236(96)90127-7
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Woodhead, J. L., Howell, B. A., Yang, Y., Harrill, A. H., Clewell, H. J. 3rd., Andersen, M. Due east., et al. (2012). An analysis of N-acetylcysteine treatment for acetaminophen overdose using a systems model of drug-induced liver injury. J. Pharmacol. Exp. Ther. 342, 529–540. doi: ten.1124/jpet.112.192930
Pubmed Abstruse | Pubmed Total Text | CrossRef Total Text | Google Scholar
Woodhead, J. L., Yang, K., Brouwer, Yard. L. R., Siler, South. Q., Stahl, S. H., Ambroso, J. L., et al. (2014). Mechanistic modeling reveals the critical knowledge gaps in bile acid-mediated DILI. CPT Pharmacomet. Syst. Pharmacol. 3, e123. doi: 10.1038/psp.2014.21
Pubmed Abstract | Pubmed Total Text | CrossRef Full Text | Google Scholar
Yang, K., Woodhead, J. L., Watkins, P. B., Howell, B. A., and Brouwer, K. L. (2014). Systems pharmacology modeling predicts delayed presentation and species differences in bile acid-mediated troglitazone hepatotoxicity. Clin. Pharmacol. Ther. 96, 589–598. doi: 10.1038/clpt.2014.158
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Yang, G., Woodhead, J. L., St. Claire, R., Watkins, P. B., Siler, Due south. Q., Howell, B. A., et al. (2013). Quantitative human relationship between intracellular lithocholic acid and toxicity in rat sandwich-cultured hepatocytes: incorporation into a mechanistic model of drug-induced liver injury. Toxicologist 132:226.
Yoshikado, T., Takada, T., Yamamoto, T., Yamaji, H., Ito, One thousand., Santa, T., et al. (2011). Itraconazole-induced cholestasis: involvement of the inhibition of bile canalicular phospholipid translocator MDR3/ABCB4. Mol. Pharmacol. 79, 241–250. doi: ten.1124/mol.110.067256
Pubmed Abstract | Pubmed Full Text | CrossRef Total Text | Google Scholar
Source: https://www.frontiersin.org/articles/104964
0 Response to "What Would Happen to Someone Treated With a Drug That Inhibited the Release of Bile Salts?"
Post a Comment